

Aprobarea unui nou medicament
Orice medicament (produs cu substanta activa dozat corespunzator si produs de o companie farmaceutica) care ajunge in farmacie este supus unui proces riguros prin care se evalueaza siguranta, eficacitatea si utilitatea sa in tratarea uneia sau mai multor afectiuni medicale. In articolul de astazi vom parcurge impreuna etapele aprobarii unui medicament nou, incepand de la descoperirea moleculei si pana la prima adiministrare la pacienti dupa aprobarea sa pe piata farmaceutica.
Totul incepe din eprubeta
Orice noua substanta medicamentoasa va parcurge 2 etape importante intr-un proces ce poate dura pana la 10 ani si care costa aproximativ 1 miliard de dolari / medicament nou pus pe piata. Prima etapa se numeste etapa pre-clinica si aceasta etapa este impartita in 2 categorii distincte de teste. Intr-o prima faza, denumita „faza de laborator”, se face un screening a catorva sute sau chiar mii de molecule pentru a se descoperi posibile efecte terapeutice (ex. anti-inflamator, anti-infectios, hipoglicemiant, etc.). Aceste efecte se testeaza pe culturi de celule corespunzatoare (ex. hepatocite, adipocite, celule musculare, etc), in functie de efectul cercetat si posibilul organ tinta vizat de viitorul medicament. Odata descoperite, cateva zeci sau sute de „substante-candidat” ce indeplinesc anumite conditii (siguranta si efecte terapeutice) trec in a doua categorie de studii pre-clinice: studiile pe animale de laborator. In aceasta faza se testeaza efectele in vivo (in organism) si siguranta acestor substante in organisme vii. Aceste efecte sunt puternic dependente de sitemele folosite pentru testare (o substanta nu are acelasi efect intr-o linie celulara si/sau intr-un model animal si/sau pe subiecti umani). De aceea se folosesc aceste etape intermediare si o progresie liniara (de la un organism mai simplu catre o organizare mai complicata; linie celulara – tesut – organism) inainte de a se ajunge la studii clinice (pe subiecti umani).
Studii pe pacienti umani
De regula, din sute de substante testate pe animale, doar cateva (de obicei una) trec la urmatoarea faza, cea de studii clinice. Pentru a putea fi administrata subiectilor umani, aceasta substanta trebuie sa dovedeasca in primul rand un profil de siguranta bun si apoi, un profil terapeutic de interes in boala studiata. Studiile clinice sunt puternic reglementate si exista etape limitante puse in practica pentru a evita posibile erori ce ar putea pune in pericol sanatatea si viata pacientilor !
Fazele clinice ale testarii medicamentelor sunt in numar de 4: faza I pana la IV si, ca si in cazul fazelor preclinice, se urmeaza o progresie in ceea ce priveste testarea efectelor terapeutice si toxicitatii substantelor medicamentoase.

Fig. 1 Mii de cercetatori din domeniul medical si farmaceutic cerceteaza in continuu noi substante pe care sa le transforme in medicamente eficiente si sigure.
Faza I
Se mai numeste faza „primelor teste pe oameni” (din engl. „first in human”), fiind prima data cand substanta este administrata pacientilor umani. In aceasta faza participa doar pacienti sanatosi (in cazul majoritatii medicamentelor; in cazul unor medicamente oncologice care sunt urgente e posibil sa participe direct pacienti) si se determina doza optima administrata si siguranta substantei farmaceutice. De obicei, in aceasta prima faza participa cateva zeci de pacienti si se testeaza doze crescande pentru a se determina doza optima ce se poate administra cu un profil bun de siguranta. Se urmaresc aici si aparitia primelor posibile reactii adverse si efecte toxice ce ar putea opri trecerea substantei in faze superioare de testare.
Faza II
In faza II se recruteaza un numar mai mare de participanti (cateva sute) si se urmareste pentru prima data eficacitatea substantei in tratarea bolii tintite. In aceasta faza vor participa pacientii ce sufera de boala pentru care se testeaza medicamentul. Se pot face si teste genetice care sa ajute la intelegerea mai buna a metabolizarii medicamentului de catre organism. In aceasta faza se opresc majoritatea substantelor, acestea neavand efectele terapeutice dorite sau un profil de siguranta satisfacator. Aproximativ 1 din 5 substante trece cu bine de faza II in fara III. Cand se face designul studiului, substanta medicamentoasa se va administra in mod randomizat (alegerea la intamplare, de obicei de catre un software computerizat) unui anumit pacient. Se va compara grupul pacientilor care primesc substanta fie versus unui grup ce primeste placebo (forma farmaceutica fara nicio substanta medicamentoasa) sau tratamentul standard in acea boala (pentru a se compara fata de cea mai buna optiune terapeutica). Food and Drug Administration (Organizatia care se ocupa de aprobarea medicamentelor in SUA) recomanda ca toate noile substante sa se testeze in comparatie cu un placebo pentru a demonstra efectul terapeutic intrinsec.
Faza III
Daca o substanta terapeutica a ajuns in faza III inseamna ca are un potential bun sa ajunga pe piata farma. Are un profil bun de siguranta pe cei cateva sute de subiecti testati si, in plus, are si efecte terapeutice acceptabile (sau chiar foarte bune). Aici, se creste brusc numarul de pacienti care au boala, ajungandu-se la testarea substanei pe mii de pacienti in studii mari, numite studii de cohorta. Se alcatuiesc mai multe echipe de cercetare clinica (formate din medici din clinici) si se studiaza medicamentul in mai multe orase, tari si continente simultan (studii multi-centrice). Aceste studii vor aduce rezultate foarte importante legate de profilul de eficacitate si de siguranta a medicamentului in conditii clinice, reale de tratare a pacientilor. 1 din 2 substante care sunt testate in faza III nu vor fi aprobate pentru utilizare. In aceasta faza se cheltuie si cei mai multi bani, fiind estimat ca un sponsor mare (companie farmaceutica puternica) cheltuie in medie in jur de 450 de milioane de dolari pentru un medicament aflat in faza III si un sponsor mic (companie farmaceutica mica) cheltuie in jur de 15 milioane pentru fiecare medicament aflat in aceasta faza de studii clinice. Aceste cheltuieli depind de numarul de centre incluse in cercetare, numarul de pacienti recrutati, durata de urmarire a pacientilor (in unele cazuri, in boli cronice, pacientii trebuie urmariti mult timp pentru a se observa un efect terapeutic). Cand un medicament trece cu bine de faza III, acesta este, de regula, aprobat pentru a fi utilizat la pacienti.
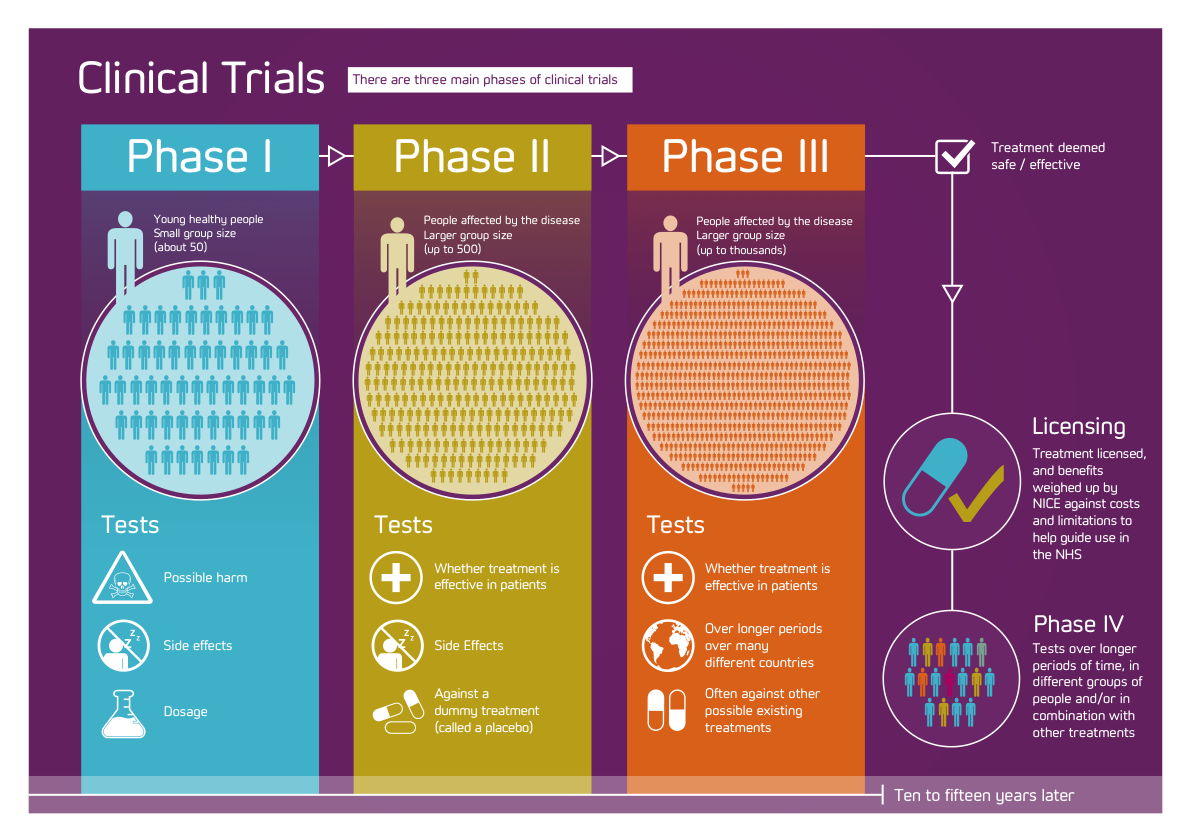
Fig.2 Fazele studiilor clinice, numarul de persoane ce participa si obiectivele urmarite. Reprodus din: https://www.dementiablog.org/the-global-clinical-trials-fund-drugs-and-dummies/.
Faza IV
Dupa ce medicamentul este pus pe piata, se urmareste in continuare profilul sau de siguranta si eficacitatea sa. Acum medicamentul este administrat la un numar mult mai mare de pacienti decat in studiile clinice si e posibil sa apara reactii adverse noi, nesemnalate anterior. Aceasta faza se numeste faza post-marketing sau faza de „farmacovigilenta”, medicamentul fiind urmarit cel putin 2 ani dupa punerea pe piata sau, in unele cazuti pe toata durata vietii sale.
In anumite conditii, unele medicamente deja aprobate pot fi re-aprobate pentru alte boli, acestea parcugand un drum mai scurt in studii clinice (de vreme ce siguranta se cunoaste deja) si fiind mai rapid accesibile pacientilor.
Toate aceste etape ale studiilor clinice sunt puternic reglementate si urmarite de organizatii de profil (EMEA pentru Europa si FDA pentru SUA fiind primele 2 din lume) care asigura punerea la dispozitie doar a medicamentelor sigure si eficiente ce pot fi utilizate de catre pacienti.
Articol original scris de Andrei Baiceanu pentru farmacist.info. Poate fi folosit cu toate drepturile de catre www.hepato.ro pentru publicare si distribuire.
Paris, Andrei Baiceanu
24.10.2016


